Hirschsprung’s disease is a congenital condition where ganglion cells of the enteric nervous system are absent in a distal portion of the bowel, most often the sigmoid colon and rectum.
Pathophysiology
The enteric nervous system (the brain of the gut) consists of two plexuses:
- Myenteric plexus (Auerbach’s plexus), primarily responsible for peristalsis
- Submucosal plexus (Meissner’s plexus), regulates fluid secretion, blood flow and absorption
In Hirschsprung’s disease, the ganglion cells of the myenteric plexus and submucosal plexus are absent. During fetal development, these cells start higher in the gastrointestinal tract and gradually migrate down to the distal colon and rectum. Hirschsprung’s occurs when the ganglion cells do not migrate all the way down the colon, and a section is left without these cells.
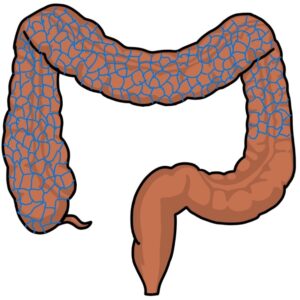
The length of the colon without innervation varies between patients from a small area of the rectum to the entire colon. When the entire colon is affected, this is called total colonic aganglionosis.
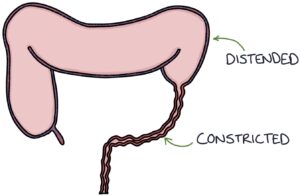
The aganglionic section of the colon does not relax, causing it to become constricted, leading to bowel obstruction. The bowel higher up (proximal) becomes dilated and overloaded.
Genetics and Associations
Many genes modify the risk of developing Hirschsprung’s. A family history greatly increases the risk. It is more common in males.
Hirschsprung’s disease usually occurs in isolation. However, it may be associated with:
- Down’s syndrome
- Waardenburg syndrome (a genetic condition causing pale blue eyes, hearing loss and patches of white skin and hair)
- Neurofibromatosis type 1
- Multiple endocrine neoplasia type II
Presentation
The presentation varies depending on the extent of the bowel that is affected.
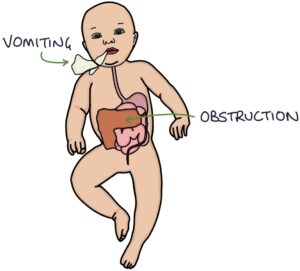
It can present shortly after birth with acute intestinal obstruction, with:
- Absent meconium
- Abdominal distension
- Vomiting
It may present with more gradually developing symptoms of:
- Delay in passing meconium (more than 24 hours)
- Chronic constipation (starting at birth)
- Abdominal pain and distention
- Vomiting
- Poor weight gain and failure to thrive
Management
Rectal biopsy is used to confirm the diagnosis. The bowel histology demonstrates an absence of ganglion cells.
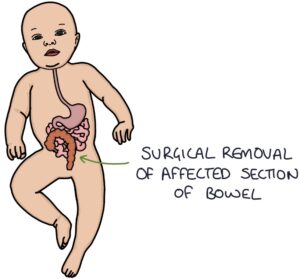
Definitive management is by surgical removal of the aganglionic section of the bowel. Most patients will live a normal life after surgery, although they may have long-term bowel function disturbances and incontinence.
Hirschsprung-Associated Enterocolitis
Hirschsprung-associated enterocolitis (HAEC) involves inflammation of the intestine. It occurs in around 20-30% of patients with Hirschsprung’s disease and can occur before or after surgery.
It presents with:
- Fever
- Abdominal distension
- Diarrhoea (often with blood)
- Features of sepsis
HAEC is life-threatening and can lead to toxic megacolon and perforation of the bowel. It requires urgent antibiotics, fluid resuscitation and decompression of the obstructed bowel.
Last updated February 2025
Now, head over to members.zerotofinals.com and test your knowledge of this content. Testing yourself helps identify what you missed and strengthens your understanding and retention.
![]()