Neurofibromatosis is a genetic condition that causes nerve tumours (neuromas) to develop throughout the nervous system. These tumours are benign but can cause neurological and structural problems.
Neurofibromatosis type 1 is more common than neurofibromatosis type 2. This section focuses mainly on type 1.
Neurofibromatosis Type 1 Gene
The neurofibromatosis type 1 gene is found on chromosome 17. It codes for a protein called neurofibromin, which is a tumour suppressor protein. Mutations in this gene are inherited in an autosomal dominant pattern.
Features
The diagnostic criteria for neurofibromatosis type 1 are based on the features, remembered with the “CRABBING” mnemonic:
- C – Café-au-lait spots (more than 15mm diameter is significant in adults)
- R – Relative with NF1
- A – Axillary or inguinal freckling
- BB – Bony dysplasia, such as Bowing of a long bone or sphenoid wing dysplasia
- I – Iris hamartomas (Lisch nodules), which are yellow-brown spots on the iris
- N – Neurofibromas
- G – Glioma of the optic pathway
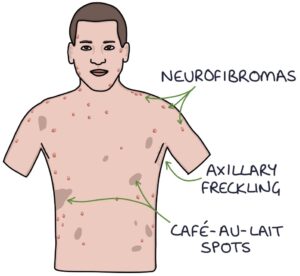
Neurofibromas may be seen on the skin. They are skin-coloured, raised nodules or papules with a smooth, regular surface. A single skin neurofibroma without other features does not indicate neurofibromatosis. Two or more are significant. A plexiform neurofibroma is a larger, irregular, complex neurofibroma containing multiple cell types. A single plexiform neurofibroma is significant.
Management
Diagnosis is based on the diagnostic criteria. Genetic testing can be helpful.
There is no treatment for the underlying disease process. Management involves monitoring, managing symptoms and treating complications.
Complications
- Migraines
- Epilepsy
- Renal artery stenosis, causing hypertension
- Learning disability
- Behavioural problems (e.g., ADHD)
- Scoliosis of the spine
- Vision loss (secondary to optic nerve gliomas)
- Malignant peripheral nerve sheath tumours
- Gastrointestinal stromal tumour (a type of sarcoma)
- Brain tumours
- Spinal cord tumours with associated neurology (e.g., paraplegia)
- Increased risk of cancer (e.g., breast cancer and leukaemia)
TOM TIP: The two unique complications worth remembering for NF1 are malignant peripheral nerve sheath (MPNST) and gastrointestinal stromal tumours (GIST).
Neurofibromatosis Type 2
The neurofibromatosis type 2 gene is found on chromosome 22. It codes for a protein called merlin, a tumour suppressor protein important in Schwann cells. Schwann cells provide the myelin sheath that surrounds neurones of the peripheral nervous system. Mutations in this gene lead to schwannomas (benign tumours of the Schwann cells). Inheritance is also autosomal dominant.
Neurofibromatosis type 2 is particularly associated with acoustic neuromas, which are tumours of the auditory nerve that innervates the inner ear.
Surgery can be used to resect the tumours, although there is a risk of permanent nerve damage.
TOM TIP: An exam patient with bilateral acoustic neuromas almost certainly has neurofibromatosis type 2.
Last updated October 2023
Now, head over to members.zerotofinals.com and test your knowledge of this content. Testing yourself helps identify what you missed and strengthens your understanding and retention.
![]()