Thalassaemia is caused by a genetic defect in the protein chains that make up haemoglobin.
Normal haemoglobin consists of two alpha-globin and two beta-globin chains.
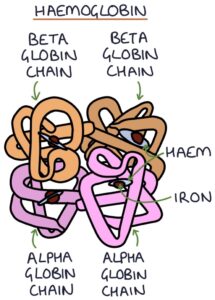
Defects in alpha-globin chains lead to alpha thalassaemia. Defects in the beta-globin chains lead to beta thalassaemia.
Both conditions are autosomal recessive.
Ineffective red blood cell production (erythropoiesis) leads to anaemia. The red blood cells are fragile and break down easily, causing haemolytic anaemia. Splenomegaly results from increased clearance of abnormal cells and extramedullary haemopoiesis (red blood cell production outside the bone marrow).
All pregnant women in the UK are offered a screening test for thalassaemia when booking with a midwife.
Features
The severity of features depends on the type. Features include:
- Microcytic anaemia (low mean corpuscular volume)
- Fatigue
- Pallor
- Jaundice
- Gallstones
- Splenomegaly
- Poor growth and development
Investigations
Microcytic anaemia (low mean corpuscular volume) is seen on a full blood count.
Haemoglobin electrophoresis is used to diagnose globin abnormalities.
DNA testing can be used to look for the genetic abnormality.
Iron Overload
Iron overload may occur in thalassaemia due to:
- Increased iron absorption in the gastrointestinal tract
- Blood transfusions
Iron overload in thalassaemia can cause symptoms and complications of:
- Liver cirrhosis
- Hypogonadism
- Hypothyroidism
- Heart failure
- Diabetes
- Osteoporosis
Ferritin levels are monitored. Raised ferritin suggests iron overload. Management involves the selective use of blood transfusions and iron chelation.
Alpha Thalassaemia
Defects in the alpha-globin chains cause alpha thalassaemia. The genes that code for alpha-globin are found on chromosome 16. The severity of symptoms varies depending on the type and number of genetic defects, ranging from entirely asymptomatic as a carrier, to moderate anaemia (haemoglobin H disease), to intrauterine death due to severe fetal anaemia (alpha thalassaemia major).
Management involves:
- Monitoring
- Blood transfusions
- Splenectomy may be performed
- Bone marrow transplant can be curative
Beta Thalassaemia
Defects in beta-globin chains cause beta thalassaemia. The gene coding for this protein is on chromosome 11. Based on the gene abnormalities, beta thalassaemia can be split into three types:
- Thalassaemia minor
- Thalassaemia intermedia
- Thalassaemia major
Thalassaemia Minor
Patients with beta thalassaemia minor (also called thalassaemia trait) are carriers of an abnormally functioning beta-globin gene. They have one abnormal and one normal gene.
Thalassaemia minor causes mild microcytic anaemia and usually only requires monitoring.
Thalassaemia Intermedia
Patients with beta thalassaemia intermedia have two abnormal copies of the beta-globin gene. The severity of symptoms depends on the mutation.
Thalassaemia intermedia causes more significant microcytic anaemia. Patients require monitoring and may need occasional blood transfusions. They may require iron chelation to prevent iron overload.
Thalassaemia Major
Patients with beta thalassaemia major have two severely abnormal copies of the beta-globin gene. Patients present in the first two years of life with anaemia, jaundice and failure to thrive.
The bone marrow expands to produce extra red blood cells to compensate for the chronic anaemia, which can cause a change of physical appearance. Abnormal features relating to bone changes include:
- Frontal bossing (prominent forehead)
- Enlarged maxilla (prominent cheekbones)
- Depressed nasal bridge (flat nose)
- Protruding upper teeth
Management may involve:
- Regular transfusions
- Iron chelation
- Splenectomy
- Bone marrow transplantation (can be curative but involves significant risks)
Last updated March 2026
Now, head over to members.zerotofinals.com and test your knowledge of this content. Testing yourself helps identify what you missed and strengthens your understanding and retention.
![]()