Systemic sclerosis is an autoimmune connective tissue disease involving inflammation and fibrosis (hardening or scarring) of the connective tissues, skin and internal organs. The cause is unclear.
Scleroderma translates directly to the hardening of the skin. The terms systemic sclerosis and scleroderma are often used interchangeably. Most patients with scleroderma have systemic sclerosis. However, a localised version of scleroderma only affects the skin.
There are two main patterns of disease in systemic sclerosis:
- Limited cutaneous systemic sclerosis
- Diffuse cutaneous systemic sclerosis
Limited Cutaneous Systemic Sclerosis
Limited cutaneous systemic sclerosis is the more limited version of systemic sclerosis. It used to be called CREST syndrome. CREST forms a mnemonic for remembering the features of limited cutaneous systemic sclerosis:
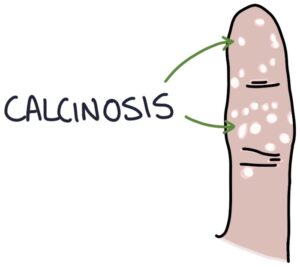
- C – Calcinosis
- R – Raynaud’s phenomenon
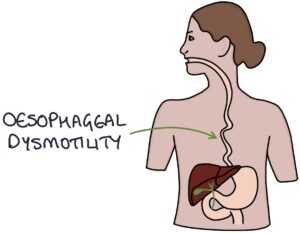
- E – oEsophageal dysmotility
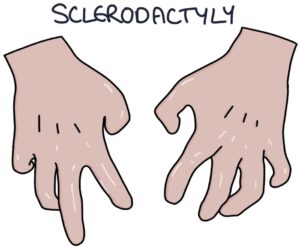
- S – Sclerodactyly
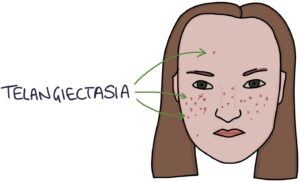
- T – Telangiectasia
Diffuse Cutaneous Systemic Sclerosis
Diffuse cutaneous systemic sclerosis includes the CREST features and also affects internal organs, causing:
- Cardiovascular problems (e.g., hypertension and coronary artery disease)
- Lung problems (e.g., pulmonary hypertension and pulmonary fibrosis)
- Kidney problems (e.g., glomerulonephritis and scleroderma renal crisis)
Features
Scleroderma refers to the hardening of the skin, giving the appearance of shiny, tight skin without the normal skin folds. These changes are most notable on the hands and face.
Sclerodactyly describes the skin changes in the hands. Skin tightening around the joints restricts the range of motion and reduces function. The fat pads on the fingers are lost. The skin can break and ulcerate.
Telangiectasia refers to dilated blood vessels in the skin measuring less than 1mm in diameter.
Calcinosis refers to calcium deposits under the skin, most commonly found on the fingertips.
Oesophageal dysmotility is caused by atrophy and dysfunction of the smooth muscle, as well as fibrosis of the oesophagus. It causes swallowing difficulties, chest pain, acid reflux and oesophagitis.
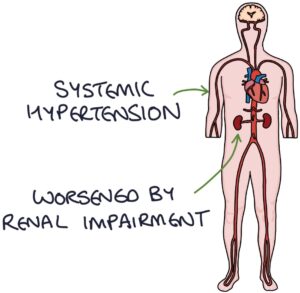
Systemic and pulmonary hypertension is caused by connective tissue dysfunction in the systemic and pulmonary arterial systems. Systemic hypertension can be worsened by renal impairment.
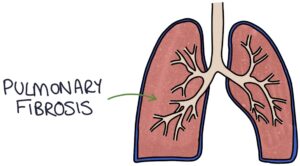
Pulmonary fibrosis occurs in severe systemic sclerosis, with a gradual onset of dry cough and shortness of breath.
Scleroderma renal crisis is a medical emergency with severe hypertension and renal failure.
Raynaud’s Phenomena
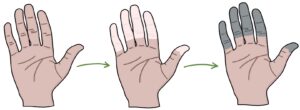
Raynaud’s phenomenon is where the fingertips change colour in response to even mildly cold triggers (e.g., opening the fridge). It is caused by vasoconstriction of the vessels supplying the fingers. The typical pattern is:
- First white, due to vasoconstriction
- Then blue, due to cyanosis
- Then red, due to reperfusion and hyperaemia
Raynaud’s disease is where Raynaud’s phenomenon occurs without an associated systemic disease. It is idiopathic and makes up 80-90% of patients with Raynaud’s phenomenon.
Systemic sclerosis is the most important secondary cause. It is less commonly associated with SLE.
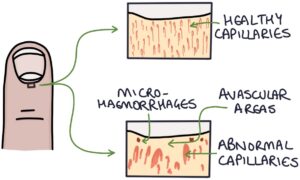
Nailfold capillaroscopy is a technique to magnify and examine the peripheral capillaries where the skin meets the base of the fingernail (the nail fold). Abnormal capillaries, avascular areas and micro-haemorrhages suggest systemic sclerosis. Patients with Raynaud’s disease (without systemic sclerosis) have normal nailfold capillaries.
Treatment options for Raynaud’s include:
- Keeping the hands warm (e.g., gloves and hand warmers)
- Calcium channel blockers (e.g., nifedipine)
- Other specialist drugs include losartan, ACE inhibitors, sildenafil and fluoxetine
Beta blockers can worsen the symptoms of Raynaud’s.
Autoantibodies
Antinuclear antibodies (ANA) are positive in most patients with systemic sclerosis. They are non-specific.
Anti-centromere antibodies are most associated with limited cutaneous systemic sclerosis.
Anti-Scl-70 antibodies are most associated with diffuse cutaneous systemic sclerosis and more severe disease.
Diagnosis
The American College of Rheumatology / European League Against Rheumatism (ACR/EULAR) classification criteria from 2013 can be used to make the diagnosis. This takes into account the clinical features, antibodies and nailfold capillaroscopy.
Management
DMARDs (e.g., methotrexate) and biologic therapies (e.g., rituximab) are options in diffuse disease. Steroids may be considered but are associated with an increased risk of scleroderma renal crisis.
Non-medical management involves:
- Avoiding smoking
- Gentle skin stretching to maintain the range of motion
- Regular emollients
- Avoiding cold triggers for Raynaud’s
- Physiotherapy to help maintain healthy joints
- Occupational therapy for adaptations to daily living to cope with limitations
Medical management focuses on treating symptoms and complications:
- Nifedipine is usually first-line for Raynaud’s phenomenon
- Proton pump inhibitors (e.g., omeprazole or lansoprazole) for acid reflux
- Prokinetic medications (e.g., metoclopramide) for gastrointestinal symptoms
- Analgesia for joint pain
- Antibiotics for skin infections
- Antihypertensives (e.g., ACE inhibitors) to treat hypertension and scleroderma renal crisis
- Endothelin receptor antagonists (e.g., bosentan) may be used for pulmonary hypertension and digital ulcers
- Sildenafil may be used for pulmonary hypertension and digital ulcers associated with Raynaud’s
- Intravenous iloprost may be used for digital ulcers
- Oxygen where required for pulmonary fibrosis
- Stem cell transplantation may be considered in rapidly progressing severe disease but carries significant risks
Last updated August 2023