Acromegaly is the result of excessive growth hormone (GH).
Pathophysiology
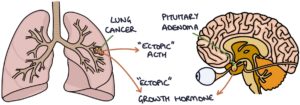
Growth hormone is produced by the anterior pituitary gland. The most common cause of unregulated growth hormone secretion is a pituitary adenoma. This adenoma can be microscopic or a significantly sized tumour that causes compression of local structures.
Very rarely, acromegaly can also be secondary to cancer, such as lung or pancreatic cancer, with a tumour that secretes ectopic growth hormone-releasing hormone (GHRH) or growth hormone (GH). This is a paraneoplastic syndrome, meaning that it occurs alongside (para-) the neoplasm (tumour).
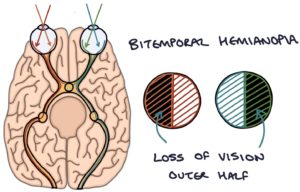
The optic chiasm sits just above the pituitary gland. The optic chiasm is where the optic nerves from the eyes cross over to the opposite side of the head before traveling to the visual cortex in the occipital lobe. A pituitary tumour of sufficient size can press on the optic chiasm. Pressure on the optic chiasm leads to a stereotypical bitemporal hemianopia visual field defect. This describes a loss of the outer half of the vision in both eyes. The inner half of the vision is spared, as this does not cross at the optic chiasm and stays on the same side of the head.
Presentation
A space-occupying pituitary tumour can cause:
- Headaches
- Visual field defect (bitemporal hemianopia)
Excess growth hormone causes tissue growth:
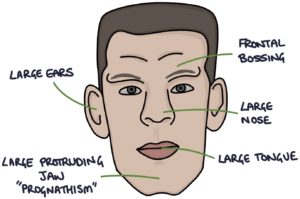
- Prominent forehead and brow (frontal bossing)
- Coarse, sweaty skin
- Large nose
- Large tongue (macroglossia)
- Large hands and feet
- Large protruding jaw (prognathism)
Additional features include:
- Hypertrophic heart
- Hypertension
- Type 2 diabetes
- Carpal tunnel syndrome
- Arthritis
- Colorectal cancer
TOM TIP: When preparing for the PACES exam, the link between bilateral carpal tunnel syndrome and acromegaly came up several times. Cases would present a patient with symptoms of bilateral carpal tunnel syndrome. The challenge was not only to diagnose carpal tunnel syndrome but also to identify the features of the underlying cause. Whenever you see a patient in an OSCE station, and you make a diagnosis, ask yourself whether that diagnosis might have an underlying cause and look for features of that cause.
Investigations
Insulin-like growth factor-1 (IGF-1) can be tested on a blood sample. It indicates the growth hormone level and is raised in acromegaly. Testing growth hormone directly is unreliable as it fluctuates throughout the day.
The growth hormone suppression test involves consuming a 75g glucose drink with growth hormone tested at baseline and 2 hours following the drink. The glucose should suppress the growth hormone level. Failure to suppress growth hormone indicates acromegaly.
MRI of the pituitary is used to diagnose a pituitary adenoma, although it may be too small to see on the scan.
Treatment
Trans-sphenoidal surgery, through the nose and sphenoid bone, to remove the pituitary tumour is the definitive treatment of acromegaly secondary to pituitary adenomas. Where acromegaly is caused by ectopic hormones from pancreatic or lung cancer, treatment ideally involves surgical removal of these tumours.
![]()
Radiotherapy may be used as part of treatment.
Medical options for reducing growth hormone are used in patients where surgery is not suitable:
- Pegvisomant is a growth hormone receptor antagonist given daily by a subcutaneous injection
- Somatostatin analogues (e.g., octreotide) block growth hormone release
- Dopamine agonists (e.g., bromocriptine) block growth hormone release
Somatostatin is also known as growth hormone-inhibiting hormone. It is normally secreted by the brain, gastro-intestinal tract and pancreas in response to complex triggers. One of the functions of somatostatin is to block growth hormone release from the pituitary gland.
Dopamine also has an inhibitory effect on GH release. However, it is weaker than somatostatin.
Last updated March 2023